
Artificial Intelligence and Machine Learning get a lot of attention these days. Machine Learning (ML) is a mathematical technique that uses empirical data to generate an algorithm that can predict or make decisions on new data. But the old adage of “garbage-in, garbage-out” especially applies in ML, so it is important to understand the data that is used to generate these models, and thoroughly question the answers the models give us. In general, ML should be used to narrow decision making, or reduce noise in data so that the task of analysis is lessened. In some well-determined cases, decisions can be made automatically (i.e., facial recognition). As a science and technology company in the biomedical space, BioTeam is interested in better understanding how ML can be used to speed drug discovery. Here, we show an extremely relevant ML exercise we have been working on for the last couple of weeks that is a naive attempt to identify a drug that could be active against SARS-CoV-2 (COV).
Researchers around the world are racing to find treatments and vaccines to eradicate COVID-19. Researchers can not leave any stone unturned. How do you search through mountains of compounds to find effective treatments? One answer is using ML to search traditional and non-traditional places for a treatment. Could there be an approved drug already out there that could be used as an “off-label” for treatment COVID-19? While we are not offering scientific insight into this problem here, we do demonstrate how biomedical research organizations and pharmaceutical companies might be able to speed the discovery of treatment options using ML.
These results are preliminary at best and need to be thoroughly explored and peer reviewed before any conclusions or medically-relevant actions can be taken. This exercise is about ML, not COV.

This article has been created and written by Fernanda Foertter, a Senior Scientific Consultant at BioTeam. Prior to joining BioTeam, she was the Global Alliance Manager for Genomics HPC and AI at NVIDIA Corporation, and led research and training efforts at Oak Ridge National Lab. She has a background in high performance computing (HPC), artificial intelligence (AI), molecular dynamics, computational chemistry, agricultural genomics and generally helping people reach their scientific and research goals using computing.
Find her on Twitter @hpcprogrammer
Something Old Becomes Something New
In preparation for a webinar on April 29th we attempted to replicate the work done by Jonathan Stokes, et. al. at the Broad Institute in Cambridge, MA. They developed a model to predict new antibiotics from known drugs. Stokes leveraged data from the Drug Repurposing Hub to demonstrate how Halicin, a drug that showed poor results for the treatment of diabetes, could act as an antibacterial molecule. The computational work was followed by in-vivo experiments that confirmed Halicin’s broad-spectrum antibiotic activity which was able to treat both Clostridium difficile and pan-resistant Acinetobacter baumannii infections in mice. Fascinating, groundbreaking work.
The ML framework Dr. Stokes’ team used ChemProp, was developed by The MIT Computer Science and Artificial Intelligence Laboratory and uses ensemble models to predict molecular properties using a method called message passing neural network. The framework can be trained on datasets containing molecules with known property values. These models can then be used to predict properties for new molecules.
Stokes JM, Yang K, Swanson K, et al. A Deep Learning Approach to Antibiotic Discovery. Cell. 2020;180(4):688–702.e13. doi:10.1016/j.cell.2020.01.021
ChemProp: Machine Learning for Molecular Property Prediction, http://chemprop.csail.mit.edu/
The framework repository includes a few pre-built models, two of which were trained on drugs known to be active on a COV protein called 3-cytokine-like protease (3CLpro). The target, 3CLpro, is a non-structural protein that is a key enzyme in the viral life cycle of the COV involved in the maturation and activation of key viral replication proteins. Finding a drug that disrupts this protease would interfere with the viruses’ ability to replicate. ChemProp uses a binary feature approach to codify whether the training set of drugs were known to be active (1) with other drugs assumed to be inactive (0). ChemProp included two models they generated based on data from a lab assay (PubChem assay AID1706) that measured the activity of 290K compounds for their ability to inhibit 3CLpro in vitro, and discovered 405 active compounds.
As we did our work, our lead scientist had just taken their asthma medication, montelukast (Singulair®*), so, out of pure curiosity, we used its molecular representation as the first input to play with the framework.
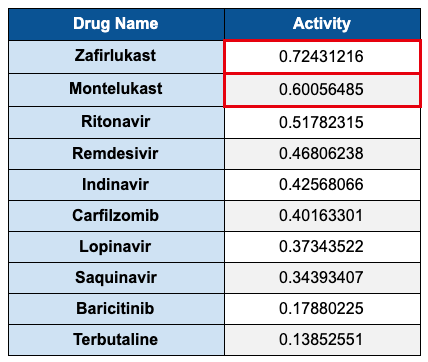
The model predicted an activity probability of 0.60. We were not sure if this was good or bad, so we found a number of antivirals from literature and tested them also. One by one those numbers came below the montelukast score (see Table 1 below).
At this point the SARS-CoV model is looking curious, even questionable. How does an asthma drug have a higher prediction than antivirals with known activity at this protein site? As mentioned earlier, it is important to always deeply question the results that come out of ML inference exercises, so we set out to prove these results wrong.
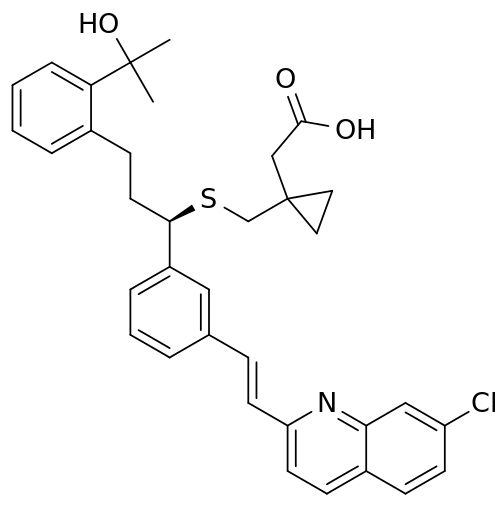
SMILES structure for Montelukast
Taking a deeper dive
A single result without context is meaningless, so as part of this exercise we wanted to understand how the model was created. We needed to ask more questions:
- How many drugs were considered positive (active) in the training set?
- What is the total number of drugs in the training set?
- Where did the training set come from?
- Are the drugs skewed toward a particular family of drugs?
We explored the ChemProp GitHub repository and the available datasets they included and considered retraining the model. The training set is indeed skewed where the vast majority of the drugs, 99.9%, are coded negative for activity at 3CLpro. This means the model will be excellent at predicting what doesn’t have activity at 3CLpro, not what would be active. But the authors of ChemProp knew this would be common in drug-related data today and developed an approach that can yield a more balanced model by exposing each batch of samples with an equal number of positive and negative molecules. Since we had used that balanced model in our analysis, we did not need to retrain the model and run the analysis again.
Next we tested a drug in the same leukotriene receptor antagonist (LTRA) family, Accolate®** (zafirlukast). Surprisingly, the activity predicted was even higher than montelukast.
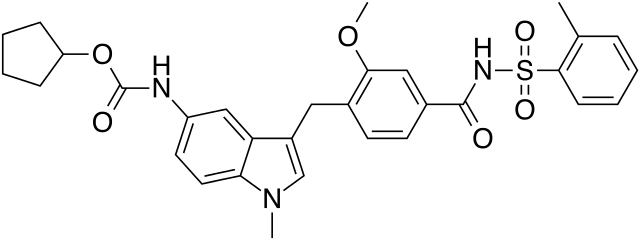
SMILES structure for Zafirlukast
What other approaches have been tried
ChemProp’s approach is one of many. There are other examples of machine learning being used to find drugs that target 3CLpro. A. Zhavoronkov, et. al., published a paper where they used a generative deep learning model to create hypothetical molecules that could target 3CLpro. They identified novel drug-like compounds that could be effective and used these hypotheticals to compare with existing drugs. Though the paper did not describe the analysis of features found across these molecules, there appeared to be some common themes. For example, the paper showed that sulfonamides were present in three of the six molecules generated, which is also present in zafirlukast. It appears that their model had identified that sulfones were an important feature in their synthetic dataset. But then again, over 100 FDA-approved drugs have sulfonamides. It’s a popular and useful theme, found in antibiotics, antivirals, anti-inflammatories and anticonvulsants. Could the training data have an over representation of these compounds? Or is this a real feature that is necessary to form the right bonds in the 3CLpro pocket?
When in doubt, go back to the literature
Our interesting findings led us to search the literature for LTRA as an antiviral. The results were thin but yielded two recent pre-prints related to coronavirus. A paper from researchers in Wuhan China where the researchers ran docking simulations that included montelukast in the list of compounds.
Out of all the compounds they found montelukast was remarkable enough to discuss specifically:
“It’s worth mentioning, anti-asthmatic drug montelukast also showed low binding energy to 3CLpro., in which lots of hydrophobic amino acids, just like Thr24, Leu27, His41, Phe140, Cys145, His163, Met165, Pro168 and His172 compose a relatively hydrophobic environment to contain the compound and stabilize its conformation. Hydrogen bonding was predicted between Asn142 and the carbonyl group of the compound (Fig. 5B).”
This means that the montelukast molecule fits and bonds in the active site of 3CLpro, potentially interfering with its function and inactivating the virus’ ability to replicate.
Another pre-print from February 2020 is this single author paper by A. Contini, a researcher out of the University of Milan, who used a simulation software called Amber to do a virtual screening of FDA-approved drugs on two SARS-CoV-2 proteins. The paper shows that montelukast had a relatively strong docking score compared to other antivirals the author tried.
These are traditional docking approaches, and very recent, not coincidentally from two major COVID-19 outbreak locations. These independent simulation (docking) studies illustrate another powerful method often used in drug discovery, that when combined with a cycle of ML inference, can refine the potential number of molecules that might have activity to a more manageable set to be tested in-vivo.
What comes next?
As shown above, there are many examples in other relevant studies suggesting that this class of drugs has antiviral properties. There have been researchers who have investigated montelukast effects on respiratory syncytial virus (RSV) and viral activity in Zika infections, and zafirlukast has been shown to inhibit NS2B-NS3 protease in West Nile virus. What machine learning did here was direct our attention to a class of drugs that has not been broadly considered to have antiviral activity, especially against Coronaviruses.
One of the exciting potential benefits of ML is to shorten the time to discovery by expanding the set of options for problem solving. In drug discovery this means identifying potential drugs from unexpected sources. Since the training dataset is what creates the model to begin with, it is critical that great care is taken to curate a training dataset that is informative enough for the problem you’re trying to solve. Then, to prove that care was taken and that the model is valid, it is equally critical to thoroughly explore the results to increase confidence that the results are actionable for further study.
While this example was not a complete scientific exploration, the extremely preliminary results were compelling enough that we have been in touch with a few collaborators to take the next step, asking them to include these compounds in their assays. Whether LTRAs have antiviral activity remains to be researched. In either case, this exercise clearly demonstrated the potential power of utilizing ML techniques in biomedicine and the type of outcomes that are possible from those efforts.
Please note that the information presented above has not been formally peer reviewed and expresses the opinions of the BioTeam.
* Singulair is a registered trademark of Merck & Co.
** Accolate is a registered trademark of AstraZeneca Pharmaceuticals LP
Neither Merck & Co., nor AstraZeneca Pharmaceuticals LP has sponsored, approved or endorsed the information, study or the findings presented in this Article.
© BioTeam, Inc. April 2020


